每日經濟新聞 2022-02-22 18:57:37
◎信達生物的PD-1藥物信迪利單抗闖關美國上市失利。該藥物在美上市溝通會上的與會專家認為,信迪利單抗的單一國家/地區臨床數據、試驗對照組和終點設計,以及與FDA的前期溝通等均未達到在美上市的要求。
◎多位創新藥行業人士則認為,信迪利單抗未能闖關成功的關鍵原因還在于“沒有滿足未滿足的臨床需求”。分析認為,對藥企來說,“從臨床試驗初始階段就考慮未滿足的臨床需求、具備國際化視野以及與FDA展開溝通都是很有必要的”。
每經記者|陳星 每經編輯|文多

圖片來源:攝圖網-401120602
今年2月10日,數以萬計的醫藥人圍觀了一場特殊的直播——信達生物(01801,HK)的PD-1藥物信迪利單抗的在美上市溝通會。作為“內卷之王”PD-1的首個闖關品種,這場溝通會的結果直接關系著國內創新藥企們的出海策略與信心。
很遺憾,5個小時的討論后,信迪利單抗以“14:1”的結果宣告闖關失利。絕大多數的與會專家一致認為,信迪利單抗的單一國家/地區臨床數據、試驗對照組和終點設計,以及與FDA(美國食品藥品監督管理局)的前期溝通等均未達到在美上市的要求。
雖然FDA的判定中存在多個細節,但多位創新藥行業人士對《每日經濟新聞》記者表示,信迪利單抗未能闖關成功的關鍵原因還在于“沒有滿足未滿足的臨床需求”。資料顯示,信迪利單抗此次申請上市的是PD-1單抗的非鱗非小細胞肺癌適應癥,在美國市場已經有四種一線治療藥物上市。其中,標準療法——“K藥”(帕博麗珠單抗)地位十分穩固。
“信迪利單抗的闖關失利并不意味著FDA對中國創新藥關上了大門,但要從這一課中學到經驗,比如把一個藥物從70分做到90分不如在一個缺醫少藥的領域研發新藥、從臨床設計開始就與國際靠攏等等,都應該是國內做藥人從這一課中學到的經驗。”一位創新藥企創始人說道。
據記者不完全統計,在信達生物身后,還有大批等待著“出海”的國內創新藥企業。包括信達生物在內,康方生物(康方生物-B,09926,HK)、百濟神州(688235,SH)都已先后向FDA遞交BLA申請(新藥上市申請)。國內的創新藥企業能否在一次次學習后“讓球最終越過球門線”,或許還得等待下一次檢驗。
圖片來源:信達生物官網截圖
2022年2月10日美東時間10點~15點,北京時間10日23點至11日4點,一場特殊的直播正在進行中。直播頁面顯示,有4萬多人圍觀了這場直播,而其中的大多數都是中國醫藥行業從業者。這場直播討論的,是信達生物信迪利單抗的美國上市申請能否獲批。
這是首個“上會”的國產PD-1單抗,也是首個基于美國以外單一國家/地區臨床數據的藥物尋求FDA批準,還是國內眾多對“出海”翹首以盼的創新藥企的探路者,其結果意義不言自明。
“14:1”——FDA ODAC(美國藥監局腫瘤藥物咨詢委員會)接近全數參與投票的專家認為,信達生物的PD-1信迪利單抗需要補充額外的臨床試驗。盡管這并非FDA給出的最終定論,但這一結果已經說明,國產PD-1登陸美國市場的時刻暫未到來。
根據直播內容及會后FDA披露的記錄文件,“僅僅擁有來自中國的臨床試驗數據,不足以說明信迪利單抗能夠滿足美國患者和美國臨床實踐的需求”,成為多位參會專家投反對票的主要原因。會議指出,信迪利單抗的ORIENT-11臨床試驗(一項評估信迪利單抗注射液或安慰劑聯合培美曲塞和鉑類,用于晚期或復發性非鱗非小細胞肺癌一線治療有效性和安全性的,隨機、雙盲、3期對照臨床研究)來自單一國家,不符合ICH E17倡導的多地區/多中心臨床試驗(MRCT)原則。
早在2017年,ICH E17(多區域臨床試驗計劃與設計的一般原則)就成為FDA的重要審評原則,該指南強調將MRCT作為全球藥物開發的首選方法。“ORIENT-11的研究人群完全由來自一個國家的亞洲患者組成,未反映美國患者的種族和民族多樣性。接受此類研究和類似研究與行業范圍內對臨床試驗公平代表性的承諾相沖突。”與會專家提出。
但單一國家或地區的臨床數據,并不是阻礙信迪利單抗在美上市的唯一因素。
按照相關監管法規,如果一款藥物的相關數據滿足美國人口和美國醫療實踐,FDA仍可采用“靈活的辦法”來評估該藥物的臨床數據。享受“監管靈活性”的前提則包括:滿足未被滿足的臨床需求;罕見病,難以開展全球多地區/中心臨床試驗;原創新藥。
對這些“靈活監管”要求,信迪利單抗也沒有滿足。一名委員在會上直言:“這項申請并沒有解決一個未被滿足的需求,我們已經有安全有效的治療方法,在總體生存率上有改善。”
此外,ORIENT-11的試驗設計不符合FDA的要求也成為討論的焦點。
有委員指出,在ORIENT-11啟動時,一線轉移性肺癌的護理標準已經發生了重大變化,即K藥聯合化療在2018年獲批用于美國的非鱗非小細胞肺癌治療,而ORIENT-11研究選擇培美曲塞聯合鉑類治療作為對照組的臨床試驗設計已經“過時”了。換言之,信迪利單抗要在美國上市,需要跟其他有相同適應癥的已上市PD-1藥物作“頭對頭”試驗,以證明自己對患者有“明顯改善”,而非僅僅證明自己的非劣性。
此外,ORIENT-11的設計終點也與美國現行腫瘤藥一線治療臨床設計的主要終點不同。
而之所以作出“過時”的臨床試驗設計,FDA認為這是信達生物方面沒有就試驗設計等與其進行溝通咨詢的結果。一名委員直指,信達和禮來方面是在試驗初步結果出爐后才找到FDA,而在此之前,FDA甚至不知道試驗正在進行中。在會議中,FDA還羅列出了與信達/禮來溝通的時間軸,以說明信達/禮來在2018年8月直接啟動了臨床試驗,但FDA在2020年4月才知曉這場試驗的進行。
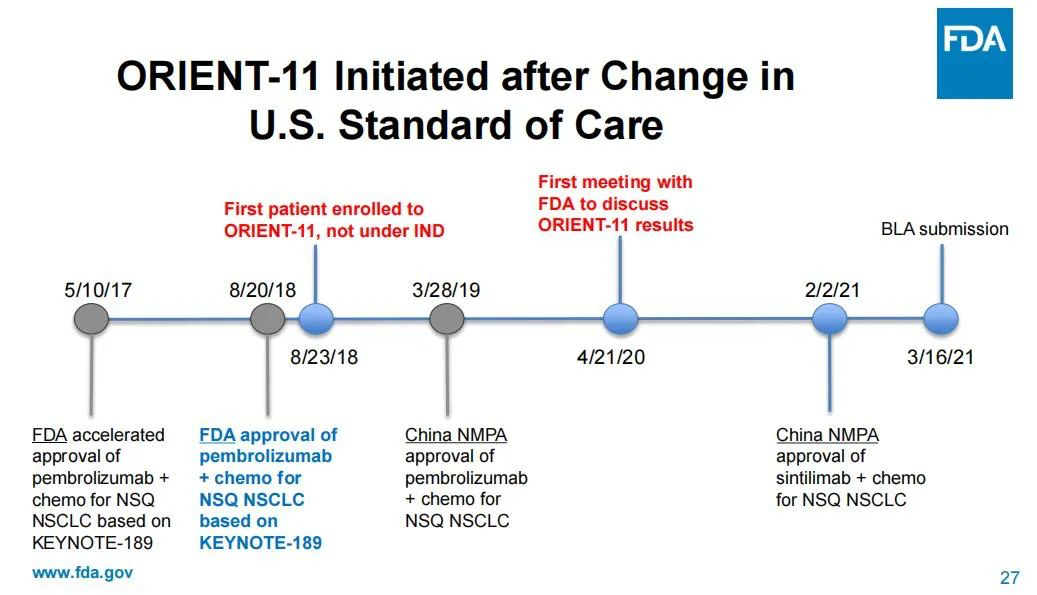
圖片來源:FDA審查資料截圖
針對該質疑,信達及合作方禮來進行了解釋。信達生物方面在接受媒體采訪時表示:“不管在臨床注冊還是上市申報階段,公司都和FDA保持了密切溝通,臨床方案也獲得了FDA認可。在2020年8月與FDA分別召開了pre-BLA關于臨床和CMC的會議,對BLA遞交資料的內容和形式進行了進一步的確認,我們并沒有收到FDA對于單一國家數據臨床申報的反對意見,或者要求必須是MRCT。”
雖然對判定的細節尚有爭議,但確定的是,信迪利單抗的美國上市節點還未到來。有市場人士預測,如果按照FDA的“頭對頭”及地區/中心要求補充臨床試驗,將是幾億美金和若干年的巨大投入,初步預估需要2000人,到2030年才會完成。即便是信達和禮來打算繼續投入,也要考慮投入與回報三思而行。
不難發現,“單一國家/地區臨床試驗數據”、“不滿足未被滿足的臨床需求”、“未與FDA充分溝通”是造成信達生物此次出海暫緩的主要原因。
其中,“不滿足未被滿足的臨床需求”成為首當其沖的關鍵因素。東吳證券醫藥行業首席分析師朱國廣認為,盡管上述FDA ODAC會議花費了大量時間討論ORIENT-11的臨床終點、對照藥物和人種多樣性等,但信迪利單抗被拒絕的核心原因仍在于“臨床需求的緊迫性不夠”。在一個有類似藥物的領域,FDA勢必用更高標準要求臨床質量。
信迪利單抗此次申請上市的是PD-1單抗的非鱗非小細胞肺癌適應癥,在美國市場已經有“O藥”“K藥”“T藥”以及西米普利單抗等治療藥物上市。以目前非鱗非小細胞肺癌的標準療法“K藥”為例,其于2014年獲批上市,2021年實現全球銷售收入171.86億美元,僅次于常年霸占銷售排行榜第一的“藥王”阿達木單抗。
在美國年銷售額超10億美元的藥物被稱為“重磅炸彈藥物”,據統計,這些藥物上市后的專利保護期也更長。
在這一背景下,作為年銷售額超百億的“超級重磅炸彈”藥物,K藥在美國市場具有更長的專利獨占期,享有絕佳的競爭環境。早在2021年10月,FDA曾拒絕了具有優先評審資格的Agenus的PD-1單抗Balstilimab的上市,理由是在K藥已經獲得宮頸癌完全批準的前提下,不適宜在基于一項單臂II試驗結果的基礎上批準該藥物的上市。
一家國內創新藥企業的創始人對《每日經濟新聞》記者表示,想要在同類適應癥上與標準療法競爭,信迪利單抗勢必要過“頭對頭”試驗這一關。同時,“聚焦未滿足的臨床需求”也以信達生物的此次嘗試為代價,得到了再一次強調。
他表示,在信達折戟的幾天之后,輝瑞的新冠口服治療藥物就獲批在國內上市。“這個對比就很有意思。輝瑞的paxlovid獲批是因為國內之前還沒有一個新冠口服治療藥,而信迪利單抗遇阻是因為美國不差這一個PD-1”。

Paxlovid 圖片來源:新華社發(輝瑞公司供圖)
在遞交上市申請之前,信達生物和禮來并非不知道美國市場已經有多個同適應癥藥物的存在,但信達和禮來希望用“低價”來叩開FDA的大門。
在此前的媒體報道中,禮來制藥方面曾表示,如果信迪利單抗成功獲批上市,與目前已經獲批上市的PD-1藥物價格相比,禮來計劃為藥物的批發采購成本提供大約40%的折扣。
低價對美國市場而言沒有吸引力嗎?在ODAC會議上,FDA明確指出,雖然FDA承認藥物成本是一個對患者有重大影響的重要社會問題,但FDA在監管決策中并不考慮定價問題。“我們不應該為了權宜之計犧牲質量,而只是為了讓球越過球門線。我們不希望因為其他原因例如成本,導致批準或驅使人們在沒有足夠數據的情況下使用藥物。”一位為信迪利單抗上市投下反對票的委員如是說。
而“低價”未能成為敲門磚的一個重要原因,在于美國的藥物定價機制與支付系統。美國藥品實行市場自由定價制度,聯邦政府不直接對藥價進行管制,而是通過批發商、藥品福利管理人等分別與藥企談判確定藥價。換言之,為了確保藥企的持續性投入和鼓勵創新研發,低價并不是美國藥品定價的最主要原則。
醫藥投資人Daniel(化名)則對《每日經濟新聞》記者表示:美國雖然有多達數十款的PD-(L)1產品,但多年來其價格體系非常穩固。如果中國的PD-1都以低價為手段進入美國市場,尤其是以大適應癥上市,可能會對美國的PD-1藥價體系、臨床資源帶來一定的影響。
換言之,“未滿足的臨床需求”是任何創新藥繞不過去的核心議題。創新藥企康寧杰瑞董事長徐霆早前接受《每日經濟新聞》記者采訪時就表示:“不管是best-in-class還是first-in-class,都不應該是目的。做藥的目的始終應該是解決臨床需求。比如阿司匹林現在在臨床上用得很好,沒必要再去做一個與阿司匹林競爭的或是新靶點同用途的藥物。”
信迪利介紹視頻 圖片來源:信達生物官網視頻截圖
信迪利單抗出海折戟后,有悲觀的聲音傳出——“FDA已經關上了對中國企業創新藥的大門”。
但在采訪中,多位受訪者一致表示,不必對信迪利單抗上市被否一事過于悲觀,“關上大門”的判斷太絕對,但信達生物的出海一課也至關重要,國內創新藥企必須從中學習經驗和教訓。
“信達只是將FDA的導向推向了頂峰,后者的要求具有持續性和普適性,但在以前的教訓中沒有得到足夠的重視。”前述創新藥企的創始人提到。他所說的教訓,是指2021年12月,萬春藥業的First-in-class新藥普那布林上市申請被FDA以臨床研究數據不夠充分的理由拒絕。
“普那布林的臨床試驗做了10多年,它的標準還是沿用的10年前的標準。普那布林做了III期試驗。但某種程度來說,它的試驗是在FDA的一個灰色區域,刺激中性粒細胞生成只做了一個III期,II期臨床做的是肺癌病人的生存獲益。對于FDA來說,如果要求松一點可能可以接受,但緊一點也可以拒絕你的上市申請。”上述創新藥企的創始人表示。
無獨有偶,按照禮來方面的表述,信迪利單抗也是在中國的III期臨床試驗數據出爐后,得知有在美國上市的可能性,所以向FDA提出上市申請。而這一點也被外界解讀為想走“捷徑”,但最終遭到了FDA的拒絕。
談到是否認為萬春藥業和信達生物是在走“捷徑”時,Daniel表示了否定。但他認為,信達生物和禮來不排除有“試一試”的心態。但很顯然,這種嘗試并沒有成功。
而另一方面,與信迪利單抗類似的是,在美國的醫療實踐中,普那布林瞄準的升白藥(提升白細胞數的藥物)市場同樣有使用多年的G-CSF(粒細胞集落刺激因子)及多種生物類似藥。換言之,普那布林沒有解決未滿足的臨床需求。
前述創始人以自己與FDA打交道的經歷表示,“FDA掌握的是匯總式的信息,它能看到的各式各樣的申請比藥企多得多。因此在FDA的考量里,不僅會考慮到現有的療法,它很可能已經看到了兩三年后的療法。FDA說你的臨床應該怎么做、對照怎么選、人群怎么設置,是基于幾年后的狀態預期。所以藥企從臨床試驗初始階段就考慮未滿足的臨床需求、具備國際化視野以及與FDA展開溝通都是很有必要的”。這也是為什么國際大藥企都在FDA辦公所在地的馬里蘭附近設辦公室的原因。
他還表示,過去美國的制藥業不把中國當成競爭對手,但“近些年差距的縮小讓FDA沒有必要給潛在的競爭對手開設綠色通道”。
據《每日經濟新聞》記者不完全統計,在信達生物身后,還有大批等待著“出海”的國內創新藥企業。包括信達生物在內,傳奇生物、康方生物、百濟神州、君實生物(688180,SH)及億帆醫藥(002019,SZ)都已先后向FDA遞交BLA申請。除信達生物外,康方生物的派安普利、百濟神州的替雷利珠以及君實生物的特瑞普利均為PD-1單抗抑制劑。

圖片來源:康方生物官網截圖
Dianel表示,君實生物的特瑞普利單抗以鼻咽癌適應癥申請上市,鼻咽癌在國外是小適應癥但存在市場需求,且目前國外的PD-1目前沒有鼻咽癌適應癥獲批。“君實的應該月底就會有消息,可以期待一下”。
“無論如何,經此一役,國內藥企想要在美國申報競爭激烈的適應癥,涵蓋美國數據、合適的對照組和國際多中心臨床都是必須滿足的要求。”Daniel補充道。
在日前舉辦的“完善醫藥創新生態推動產業健康發展”座談會上,中國醫藥創新促進會執行會長宋瑞霖談道:“國產創新藥的春天確實來了,但不是每天都是艷陽天,經常出現陰云密布天氣,中國醫藥創新仍面臨著諸多挑戰。”他表示,當前,我國醫藥創新正處于從第一階段即以資本為驅動的階段,向以臨床為導向的第二階段轉型的關鍵時期。
如需轉載請與《每日經濟新聞》報社聯系。
未經《每日經濟新聞》報社授權,嚴禁轉載或鏡像,違者必究。
讀者熱線:4008890008
特別提醒:如果我們使用了您的圖片,請作者與本站聯系索取稿酬。如您不希望作品出現在本站,可聯系我們要求撤下您的作品。
歡迎關注每日經濟新聞APP

Copyright ? 2025 每日經濟新聞報社版權所有,未經許可不得轉載使用,違者必究。
廣告熱線? 北京: 010-57613265,?上海: 021-61283008,?廣州: 020-84201861,?深圳: 0755-83520159,?成都: 028-86512112












